Metagenomic sequencing and analysis represent a groundbreaking approach in microbiology research, enabling the study of
intricate microbial communities without the difficult task of isolating and cultivating individual organisms.
Metagenomic assembly encompasses two key facets: wet-lab procedures, involving DNA extraction, library preparation, and
sequencing, and dry-lab tasks, which include read quality enhancement, DNA sequence assembly, contig/scaffold binning,
taxonomy annotation, gene prediction, and gene annotation. While assembly-independent pipelines offer avenues for
taxonomy analysis without DNA sequence assembly, the assembly step becomes indispensable when the research goal extends
beyond taxonomy analysis to encompass genome reconstruction, comprehensive gene retrieval, and organism-specific pathway
exploration.
The objective of DNA sequence assembly is clear-cut: to attain extensive contigs/scaffolds while minimizing
misassemblies and gaps, allowing for the reconstruction of complete genomes. In the context of pure bacterial culture
genome assembly, the predominant challenge lies in repetitive sequences, e.g. multiple copies of ribosomal RNA sequences
within a genome. However, in metagenomics, this process becomes exponentially challenging due to the varying sequencing
depth/coverage of microbial genomes, the presence of closely related genomes (including multiple strains of a species),
and horizontal gene transfers across phylogenetically distant genomes. Traditional short-read sequencing methods, such
as Illumina sequencing, typically yield short contigs and require extensive computational resources, thereby limiting
the advantages of DNA sequence assembly.
PacBio HiFi reads, generated through circular consensus sequencing (CCS) mode on PacBio long-read systems, have a size
of up to 15-20 kb with an accuracy exceeding 99% (Q20 or better). By harnessing the potential of HiFi reads in
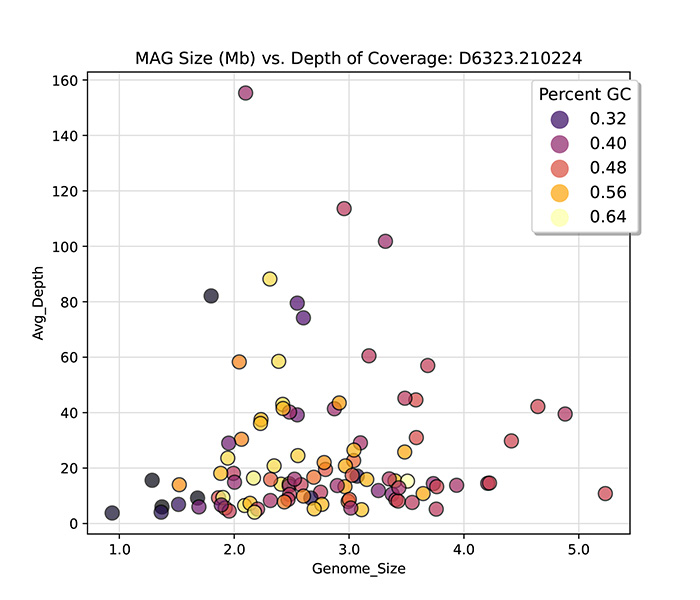
conjunction with sophisticated bioinformatics pipelines, it is now feasible to directly derive hundreds of complete or
high-quality genomes, known as metagenome-assembled genomes (MAGs), from complex metagenomic samples, such as human
fecal specimens.
At the forefront of metagenomic analysis, Zymo Research offers a Certified PacBio Provider status, ensuring the
meticulous handling of your samples. Our PacBio metagenomic sequencing and assembly service encompass the entire
workflow, ranging from DNA extraction to metagenomic assembly and comprehensive analyses. We gladly accept
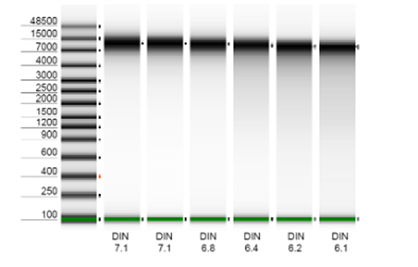
high-molecular-weight DNA extracts, or alternatively, we can perform DNA extraction when provided with raw samples. Our
use of ZymoBIOMICS DNA extraction kits guarantees impartial microbial lysis through mechanical bead beating while
preserving high molecular weight (8-15kb). Our bioinformatics pipelines encompass adapter and quality trimming, DNA
sequence assembly, binning, MAG taxonomy annotation, and taxonomy composition profiling. In the DNA sequence assembly
phase, we employ state-of-the-art algorithms optimized for metagenomic assembly with HiFi reads, as recommended by
PacBio. With us, you can have peace of mind, knowing that we will manage every aspect of your project with the utmost
care and expertise.