
SAM vs. BAM
In bioinformatics, alignment data for large numbers of aligned reads are often output as a sequence alignment and map (SAM) or binary alignment and map (BAM) file. Alignment is a common step in many bioinformatics workflows involving nucleic acid sequencing. There are many different aligners available, with the different types of aligners, their optimal applications, and how they work potentially being the subject of multiple blog posts. At their essence, aligners can be expected to take in raw sequence data in the form of a FASTQ along with a reference genome (often in the form of a FASTA file) and generate a new file containing the reads as well as the genomic location from which they originated. Most bioinformatics tools accept and expect alignment results in BAM format. From these files, with downstream bioinformatics analysis, you can compare gene expression, survey biodiversity, analyze DNA methylation, or investigate DNA-protein interaction, among many other NGS applications.
What Are SAM Files?
SAM files are a type of text file format that contains the alignment information of various sequences that are mapped against reference sequences. These files can also contain unmapped sequences. Since SAM files are a text file format, they are more readable by humans and will be used as the examples for this section.
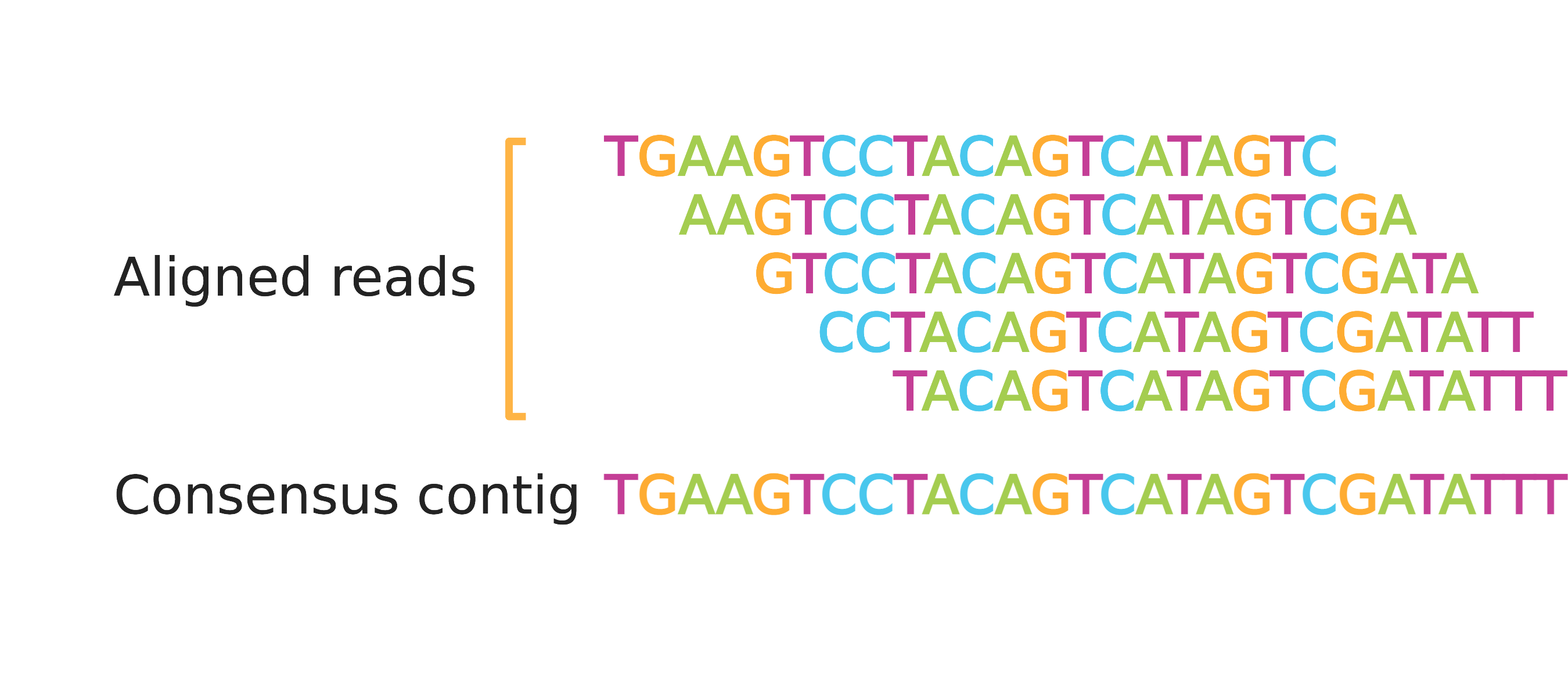
What Are BAM Files?
BAM files contain the same information as SAM files, except they are in binary file format which is not readable by humans. On the other hand, BAM files are smaller and more efficient for software to work with than SAM files, saving time and reducing costs of computation and storage. Alignment data is almost always stored in BAM files and most software that analyzes aligned reads expects to ingest data in BAM format (often with a BAM index file, to be discussed later in this post). The remainder of this piece will refer to just the BAM file for simplicity, although the data are identical between SAM and BAM files.
The two initial steps taken after the generation of a BAM file are to sort and then index it. As the reads used to generate a BAM file are (or at least should be) random regarding their positions within the genome, and BAM files often start out sorted by read identifier, if they are sorted at all. As a general rule, BAM files should be sorted as a first step to ensure that they are sorted in the way the user thinks they are. Sorting of a BAM file can be done by a few different bioinformatics applications, with Samtools and Picard being common programs for this and several other sequence analysis tasks. When sorting the BAM file, the two choices for sorting methods will be by sequence identifier, or by genomic coordinates (often referred to as location or position). The choice of method will be dependent on the downstream application, but often sorting by coordinate is the correct choice for genomic data. Most software that expects a BAM file as an input also expects that BAM file to be sorted, which is why this is often the first step in processing a BAM file.
The BAM Index File
BAM files are often accompanied by a BAM index file also known as a BAI file with a similar name. This file will always be much smaller than the BAM file and acts as a “table of contents” for the BAM file, indicating where in the BAM file a specific read or set of reads can be found. Because the location of reads within the file is likely to change with sorting, it is important to generate or regenerate the BAM index file after the companion BAM file is sorted. The creation of the BAM index file can again be done using Samtools or Picard. Most software that expects a BAM file as input also expects a companion BAI file with a similar name to be available in the same folder as the BAM.
Once a sorted BAM and its companion BAI files have been generated, there are many possible applications that can be used, depending up on the type of sequencing that was carried out and the scientific question being asked in the study.
SAM & BAM Header Lines
Header lines, as the name suggests, will be found at the start of the SAM file. They will also be found towards the beginning of the BAM file, although that is less of a concern as BAM files are not readable by humans. Header lines will always begin with an “@” symbol followed by an identifier indicating the type and subtype of the header line. Some of the most common examples may look as follows:

The first line starts with @SQ, indicating that it is identifying a reference sequence contig. The “SN” label indicates the contig name (chr14) and the “LN” label indicates contig length (107349540 bases). In this example, the only reference sequence contig was chromosome 14, but most reference genomes will have several contigs and several @SQ lines to match.
The second line starts with @PG, indicating that it describes the program used to generate the SAM file (and therefore, perform the alignment). If multiple SAM files are merged, multiple @PG lines may be present, although it is common to have only one. The “ID” tag is a unique identifier for this program with this SAM file (if multiple versions of BWA were used, the ID value would have to be modified to avoid collisions). The “PN” identifies the name of the program used (this does not need to be unique within the file) and the “VN” tag identifies the exact version of the program. Finally, the “CL” tag provides a copy of the command used to execute the alignment.
The SAM/BAM Data Line
The basic unit of data within a BAM or SAM file is the BAM or SAM line, which contains the read and alignment data for a single NGS read. There are slight differences in the encoding of data in the BAM and SAM files, but the information itself will not change between the two. Where they differ, the formatting of the SAM file will be shown, as it is human readable. BAM will always be interpreted through a computer program that can handle the different formatting and present it in a manner that is human readable. A SAM line will appear like this:
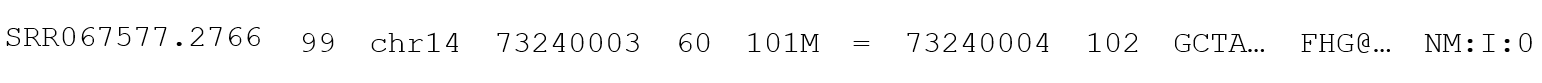
Broken into individual elements, it looks more like this:
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
99 |
chr14 |
73240003 |
60 |
101M |
= |
73240004 |
102 |
GCTA… |
FHG@… |
NM:I:0 |
The elements of this line are as follows:
- Query Name or QNAME is an identifier that is unique to the read within the file and can be used to identify any individual read. The exception to this is that mated paired-end reads will both have the same QNAME, as they are reads from the same piece of DNA, and will have to be differentiated by their direction (which can be determined from the FLAG value.
-
FLAG value, also sometimes called a flag score (a slight misnomer) is a decimal (base-10) number used to represent a binary (base-2) number with digits that represent different true/false statements pertaining to the alignment of the read. A value of zero indicates false while one indicates true.
Decimal
Binary
Exp.
Meaning
1
1
20
This is a paired read
2
10
21
This read is part of a pair that aligned properly*
4
100
22
This read was not aligned
8
1000
23
This read is part of a pair and its mate was not aligned
16
10000
24
This read aligned in the reverse direction**
32
100000
25
This read is part of a pair and its mate aligned in the reverse direction**
64
1000000
26
This read is the first in the pair (read 1)
128
10000000
27
This read is the second in pair (read 2)
256
100000000
28
The given alignment is a secondary alignment***
512
1000000000
29
Read failed quality check (such as Illumina quality filtering)
1024
10000000000
210
Read was flagged as a duplicate (such as a PCR duplicate)
2048
100000000000
211
Supplementary alignment (Exact meaning varies by aligner)
*Proper alignment indicates both reads in a pair are oriented towards one another (one forward, one reverse), are both on the same contig, and are within the expected distance from one another.
**Direction is relative to the reference sequence used for alignment
***The read had multiple potential alignments; this was one of them, but not the first choice from among them
As an example, a common FLAG value is 99, which is made up of:
64 + 32 + 2 + 1
Indicating that:
- The read is the first in pair (read 1)
- The paired-end mate of this read mapped in the reverse direction
- The read was part of a properly aligning pair
- The read was paired
One interesting property of FLAG values, caused by a value of 1 indicating that a read was paired, is that paired-end alignments should always produce odd-numbered FLAG values and odd-numbered FLAG values always indicate paired-end alignments.
- Reference Name or RNAME identifies which contig within the reference genome where the read was aligned. This value should exist in the headers in one of the @SQ lines.
- Position or POS indicates the leftmost mapping position of the first matching base within the read (see CIGAR for notes on what constitutes a match). The position value is base-1, meaning that the first letter of the reference contig is counted as position 1 (other systems may consider this position 0). A value of 0 in this field indicates a placeholder value and is not a valid position. Collectively, the reference name and position are often referred to as the genomic coordinates.
- Mapping Quality or MAPQ is a Phred-scaled confidence score indicating likelihood that the sequence was mapped correctly or incorrectly. A value of 255 for this field indicates that no probability is given and is considered a placeholder value.
-
CIGAR or concise idiosyncratic gapped alignment report string is a sequence of numbers and letters (in that order) indicating continuities or discontinuities in the alignment caused by inserted or deleted bases (or other causes for discontinuity). A typical CIGAR may appear as 5M2D5M, with the meaning of the letters explained below:
Operator
Meaning
M
Match (base may not be identical to reference, but exists in both)
D
Deletion (base exists in reference, but not read)
I
Insertion (base exists in read, but not reference)
In the above case of 5M2D5M, there were 5 bases present in both read and reference, 2 bases that were missing from the read that were present in the reference, and then another 5 bases present in both (note that there could be actual sequence mismatches where the bases differ between the reference and read in those 5 matching base stretches, as match only indicates presence).
Some additional examples:
ATCGAGCAGCCGCA
Reference
ATCGAGCAGCCGCA
Read 1
ATCGAGC---CGCA
Read 2
AT--AGCA---GCA
Read 3
The above sequences would have the following cigars:
- 14M: This sequence matches perfectly
- 7M3D4M: There are 3 bases missing in the read
- 2M2D4M3D3M: There is a 2 base and a 3 base deletion separated by 4 bases
ATCGAG--CAGCCGCA
Reference
ATCGAG--CAGCCGCA
Read 1
ATCGAGAGCAGCCGCA
Read 2
The above sequences would have the following cigars:
- 14M: This sequence matches perfectly
- 6M2I8M: There are 2 bases present in the read, but not the reference
ATCGAGCAGCCGCA
Reference
ATCGAGCAGCCGCA
Read 1
ATCTTTTTTTTGCA
Read 2
The above sequences would have the following cigars:
- 14M: This sequence matches perfectly
- 14M: The long set of “T” mismatches in the middle is still considered a match for position
- Reference Name for Mate or RNEXT is analogous to field 3 (Reference Name) and follows the same rules, except that it describes the paired-end mate of the read (if there is one). To save space, this value will be “=” if it is identical to the Reference Name value, which should be the case most often.
- Position of Mate or PNEXT is analogous to field 4 (Position) and follows the same rules as that field.
- Template Length or TLEN indicates the length of template sequence to which the read maps (this field is sometimes confused for the read length, which it is not, but will often be equal to in value). A read with multiple insertions may have a smaller template length than the read length, while a read with multiple deletions may have a template length longer than the read length. In the case of RNA or cDNA being aligned to genomic DNA reference, template length may be in the tens of thousands of bases for a short read due to the presence of an intron.
- Sequence or SEQ is the actual read sequence. Should generally follow the sequence line from the source FASTQ
- Quality String or Qual should generally follow the quality string from the source FASTQ file and be Phred-scaled.
- Predefined Tags these will be encoded tags that are predefined in the SAM/BAM file standard that give additional information on the alignment or read.